file
|
description
|
*.iso
|
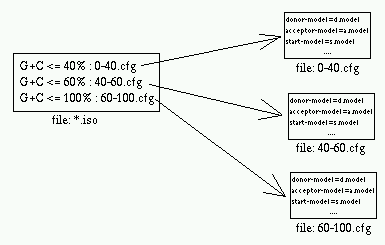
This file contains a table listing the *.cfg
file for each isochore (by G+C%).
|
*.cfg
|
This is the primary configuration file. It
Contains most of the parameters that you might want to modify.
There is one *.cfg file for each isochore.
|
transition probabilities file
|
The name of this file is specified in the
"transition-probabilities" section of the *.cfg file. It
specifies the probabilities of transitioning between particular states
in the GHMM.
|
topology definition file
|
This file defines the model topology of the
GHMM. It specifies which states have transitions to each of the
other states, which phases each signal can occur in, and several other
pieces of information which are generally not very useful to modify.
|